INTRODUCTION
Finned octopods (variously attributed to the suborder Cirrata (Cirrini) Grimpe, 1916) are generally regarded to be a sister clade to the non-finned octopods (sub-order Incirrata). These two groups have been alternatively elevated to the level of order, Cirroctopoda and Octopoda (Young 1989) or Cirromorphida and Octopodida (Sanchez et al. 2018). Cirrates retain a robust internal gladius that supports paired mantle fins, and paired cirri that flank the suckers along each of the eight arms – features shared with Jurassic and Cretaceous stem-Octobrachia including Proteroctopus ribeti Fischer et Riou, 1982 (Kruta et al. 2016), teudopseids (Fuchs & Larson 2011a), and relic vampyroteuthids (Rowe et al. 2022).
Cirrates are far less active and have a more protracted lifestyle than their incirrate relatives. To feed on small benthic invertebrates or planktonic prey, the cirrate’s webbing between the arms is extensively developed, and this coordinates with cirri in feeding (Hunt 1999, Golikov et al. 2023); their radula and salivary glands are reduced or even lost; and the ink sac and gill branchial canal are lost. The loss of the right oviduct and continuous spawning strategy reflect a longer life cycle and reduced reproductive potential (Collins & Villanueva 2006, Lindgren et al. 2012). Rapid movement is achieved in part by fins (e.g., in Grimpoteuthis; Villanueva et al. 1997) and in part by medusoid locomotion of arms and webbing (especially in Opisthoteuthis; Hunt 1999), with cirroteuthids also relying on passive drifting via the umbrella-like webbing along with gentler medusoid movements (Villanueva et al. 1997). The greatly reduced mantle musculature and smaller pallial opening reduce the effectiveness of jet propulsion (Villanueva et al. 1997, Collins & Villanueva 2006).
The classification of cirrates has been the subject of intense systematic revision over recent decades by a variety of authors, the revisions of which have operated from local to regional or global scales, and been based on a variety of molecular techniques and morphological characters and their states (Nesis 1987, Voss 1988, Voight 1997, O’Shea 1999, Piertney et al. 2003, Pardo-Gandarillas et al. 2021, Ziegler et al. 2021) (see Table 1). Consequently, nomenclature at the family and genus level has been quite unstable, aggravated by the poor condition and delicate nature of many specimens, loss or effective disintegration of types, specimen rarity, lack of genetic samples, and disagreement regarding taxonomically informative traits.
Table 1
Historical treatments of cirrate octopod genera in family level clades. Nesis (1987) is used as a starting point. Genera currently synonymized are given in parentheses below the accepted genus. Abbreviations: CirroT – Cirroteuthidae, CirroC – Cirroctopus, Grimp – Grimpoteuthididae, Lut – Luteuthididae, Opis – Opisthoteuthidae, Stauro – Stauroteuthidae
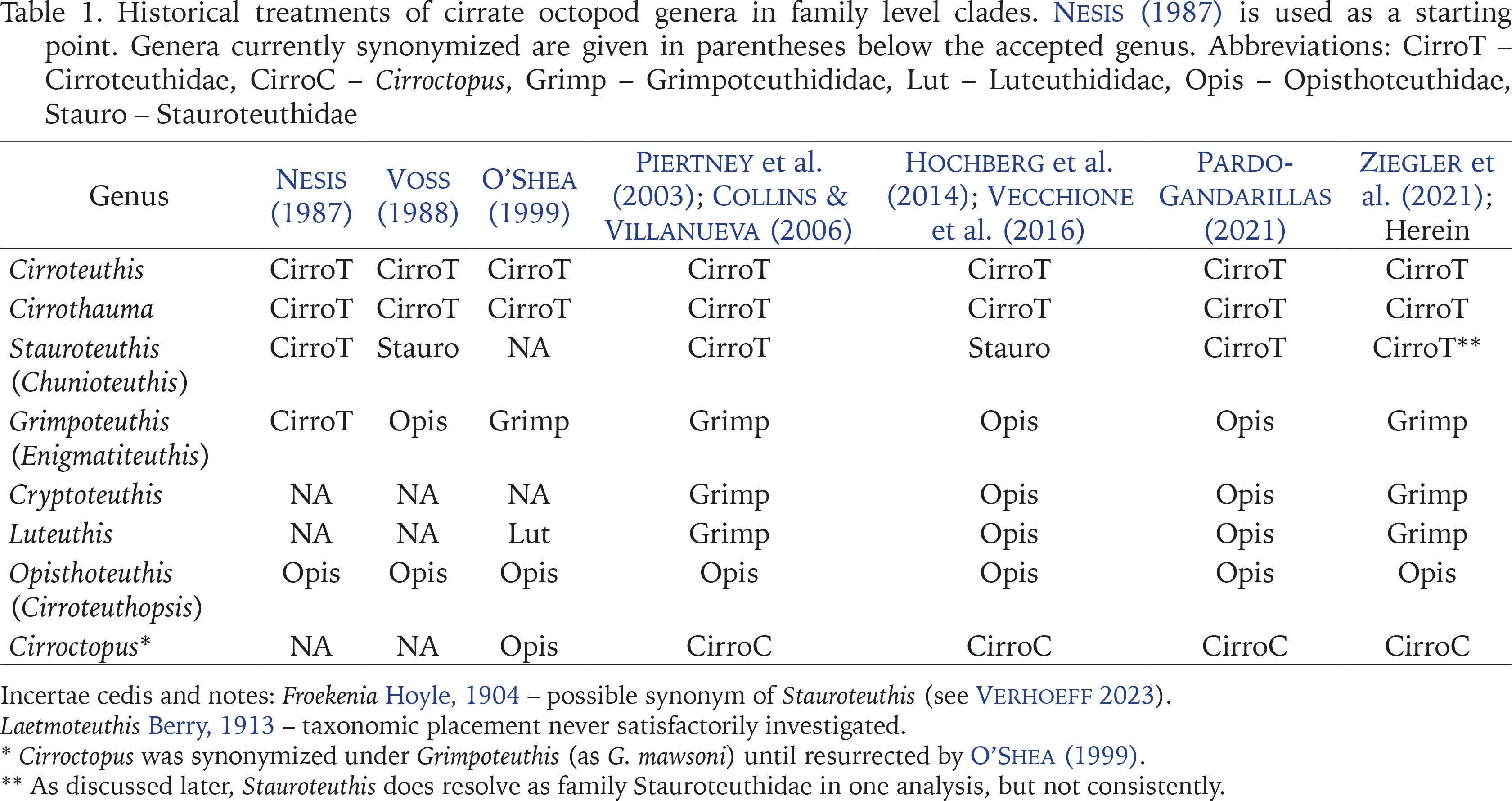
The doctoral thesis of Hudelot (2000), upon which the work of Piertney et al. (2003) is largely based, was the first comprehensive molecular appraisal of relationships among cirrate taxa, the authors electing to use the 16S mitochondrial gene for this task. The four families recognised in these studies (Cirroteuthidae Keferstein, 1866, Opisthoteuthidae Verrill, 1896, Grimpoteuthididae O’Shea, 1999, and a new family later named Cirroctopodidae Collins et Villanueva, 2006) were reviewed in the most-recent revision of the suborder by Collins & Villanueva (2006), wherein the Stauroteuthidae Grimpe, 1916 and Luteuthididae O’Shea, 1999 were considered to be junior synonyms of the Cirroteuthidae and Grimpoteuthididae, respectively. Conflicting systematic appraisals are provided by Hochberg et al. (2014) and Vecchione et al. (2016), who inexplicably recognised Stauroteuthidae but refused to recognise Grimpoteuthididae (even though the former had far less molecular support than the latter).
With more sequence data for more species and genes, including for the COI gene which has never been thoroughly investigated for the cirrates, a more comprehensive appraisal of relationships among cirrate octopods is possible. Results provide a more stable nomenclature for cirrate taxonomy, and through estimating divergence times allow for more detailed evolutionary studies. Further research that is required to better understand relationships between these deep-sea octopods is identified.
MATERIAL AND METHODS
Abbreviations: 16S – 16Svedberg ribosomal RNA (ribonucleic acid) mitochondrial gene; BI – Bayesian Inference; BIC score – Bayesian Information Criterion score; COI – cytochrome c oxidase subunit I mitochondrial gene; Ma – million years ago; ML – maximum likelihood; MP – maximum parsimony; NJ – neighbour joining; NIWA – National Institute of Water & Atmospheric Research Ltd. (New Zealand); NMNS – National Museum of Natural Science (Taichung, Taiwan).
Acquisition of molecular sequences and issues with sequence identities
All available cirrate 3'-end 16S and COI mitochondrial sequences were sourced from GenBank and BOLD databases. For whole mitochondrial sequences, aforesaid gene sequences were isolated, the 16S and COI genes being antisense to each other. Sequences, species identifications, and capture localities are detailed in Table 2 (16S) and Table 3 (COI).
Table 2
The 16S rRNA partial sequences used in this study. * – whole mitochondrial sequence. ** – sequences with regions of poor-quality (excluded from phylogenetic trees). † – ON367806 and MT725732 were both extracted from the same specimen (NIWA 95257) with the former being used herein. Abbreviations: N – north, NE – northeast, NW – northwest, S – south, SW – southwest.
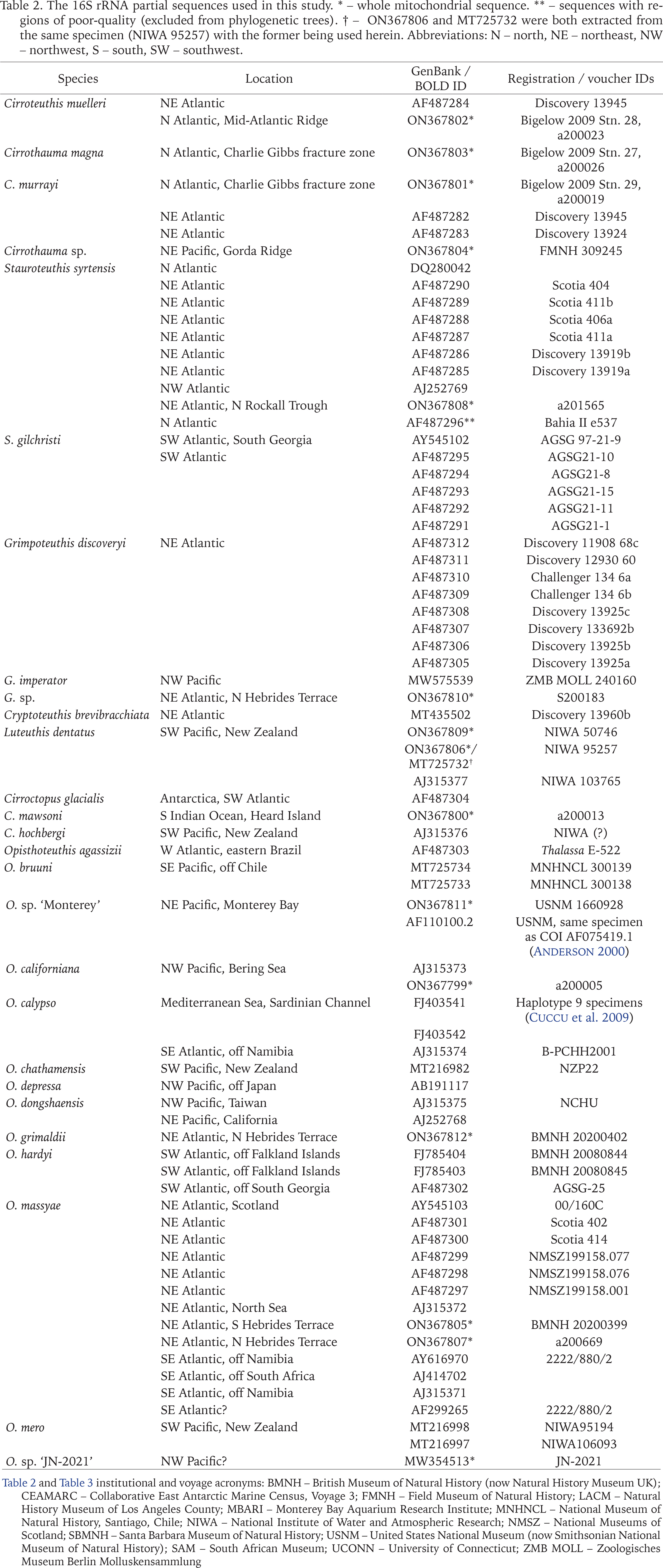
Table 3
The COI sequences used in this study. * – whole mitochondrial sequence. ** – specimens with additional data from Carlini et al. (2001). Abbreviations see Table 2
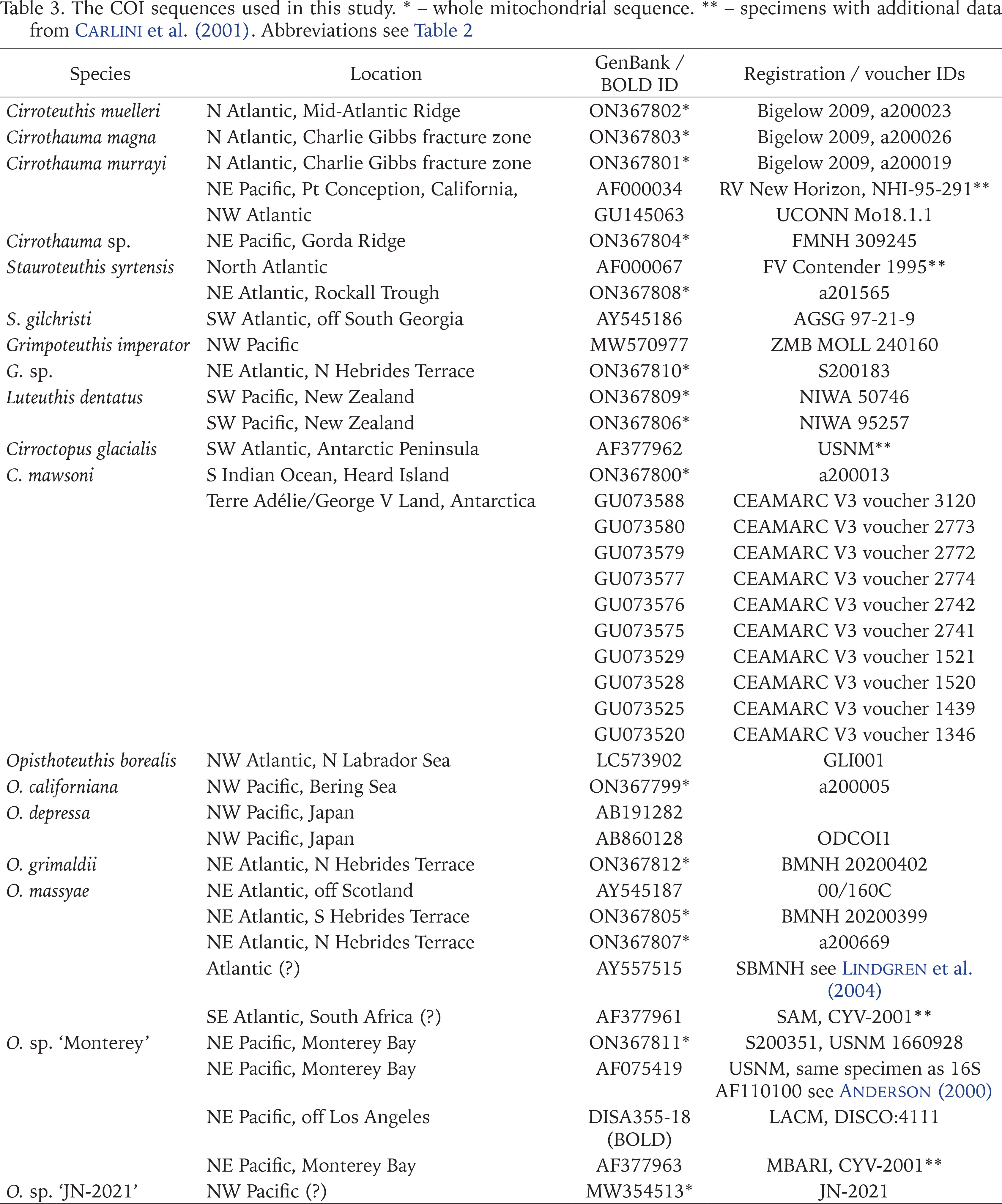
In all cases, excepting the following, existing identifications are uncritically accepted. GenBank sequence AF487303 from off eastern Brazil (RV Thalassa station E-522, 13°30.495–29.472'S, 38°38.977–37.943'W, 1,044–1,275 m, per Mincarone et al. 2014) is provisionally attributed to O. agassizii Verrill, 1883, the type species of Opisthoteuthis, because this is the only local species (Villanueva et al. 2002a). Other sequences identified to Opisthoteuthis sp. are referred to O. calypso Villanueva, Collins, Sánchez et Voss, 2002 (a) (AJ315374), O. massyae (Grimpe, 1920) (AY557515, AF377961), given their clustering with known sequences in early tree constructions, and O. dongshaensis Lu, 2010 (AJ315375) given that this is from the Opisthoteuthis tissue sample given by C.C. Lu for inclusion in Hudelot (2000) and from specimens later described by Lu (2010) as O. dongshaensis. Genbank sequence AJ315377 of Hudelot (2000) referred to L. dentatus O’Shea, 1999 was based on a sample sent by O’Shea in mid-1999 from (almost certainly) NIWA 103765 (collected March 1999) – a specimen figured by O’Shea & Lu (2002: fig. 4). Piertney et al. (2003) erroneously referred to this sample as L. shuishi O’Shea et Lu, 2002 (NMNS 002157-00088). Many COI sequences attributed to Cirroctopus sp. are herein attributed to C. mawsoni (Berry, 1917) given that they were collected from George V Land (Antarctica) close to the type locality (within the adjacent Adélie Land) (CEAMARC V3 voucher specimens; Undheim et al. 2010).
Phylogenetic inference
Alignment, trimming, and phylogenetic tree construction was conducted in MEGA X (Kumar et al. 2018). Sequencing data alignment utilised MUSCLE with settings for non-coding DNA for 16S rRNA gene data and protein coding DNA for COI. For COI, alignment was further checked by manually examining the translated amino acid sequence (ensuring no internal stop codons). Phylogenetic trees were inferred using maximum-likelihood (ML), neighbour-joining (NJ), maximum-parsimony (MP), and Bayesian inference (BI) methods. For ML trees, the substitution model and settings for rates among sites were estimated using MEGA X’s inbuilt ‘Model Selection’ tool (substitution type = nucleotide, and gaps/missing data treatment set as per trees defined later), selecting the model with the lowest (most ideal) BIC score. For each gene the final trees generally agreed with each in topology and approximate branch lengths (none being obviously more ‘accurate’), with two trees (ML and MP) combined as an illustration, and with the Bayesian inference (BI) trees illustrated separately. For both 16S and COI analyses, Octopus vulgaris Cuvier, 1797 (a generic incirrate octopus) was selected as an outgroup, as the Incirrata have been shown to be a sister group to all Cirrata (e.g., Taite et al. 2023).
All phylogenies other than Bayesian inference (ML, MP, and NJ) for both 16S and COI were tested with the bootstrap method (using 1,000 replications). Bayesian inference (BI) analyses used BEAST (Bayesian Evolutionary Analysis Sampling Trees) v1.10.4 (Suchard et al. 2018), set to run 10 million generations of MCMC (Markov Chain Monte Carlo), sampling log parameters every 1,000 generations, and discarding the first million generations as burn-in. Tracer v1.7.2 (Rambaut et al. 2018) was used to confirm the effective sampling size (ESS) and tree likelihood values, and FigTree v1.4.4 (Rambaut 2016) was used to illustrate BI trees.
16S gene analysis
For 16S rRNA gene analyses the final alignment used was 538 bp (140 bp parsimony informative, or 26.0%). ML analysis was conducted with gaps/missing data treatment set to ‘partial deletion’, and with site coverage cut-off = 90% (i.e., considering sites present in at least 90% of sequences). This analysis included 440 sites (bp positions) in the final dataset, and the HKY+G+I model was used (Hasegawa–Kishino–Yano model; discrete gamma distribution to model evolutionary rate differences among sites, 5 gamma categories, G parameter = 0.6088; allowing evolutionarily invariable sites, 44.97% sites) with BIC score = 6170.1. Per MEGAX, the initial trees for the heuristic search were obtained automatically by applying NJ (and slightly improved ‘BioNJ’) algorithms to the matrix of pairwise distances (matrix estimated using the maximum composite likelihood method) and selecting topology with a superior log-likelihood value (in this case −2587.6).
For MP analysis tree construction used Subtree-Pruning-Regrafting (SPR) as the MP search method (other tree inference options were default; initial tree obtained by random addition of sequences in 10 replicates); gaps and missing data treatment was set to ‘partial deletion’ (site coverage cut-off 90%), 440 positions in final dataset (consistency index = 0.503, retention index = 0.902, composite index = 0.498 for all sites or 0.454 for parsimony-informative sites, most parsimonious tree length = 391).
Neighbour-Joining (NJ) tree analysis used a nucleotide substitution method/model set to p-distance (including transitions and transversions), rates among sites was assumed to be uniform. All positions with < 90% site coverage were eliminated), 440 positions in final dataset (the optimal NJ tree had a sum of branch lengths = 0.770).
Bayesian Inference (BI) tree analysis used the same HKY+G+I model as the ML analysis, using 4 gamma categories, partitioning into three codon positions, and using UPGMA as a starting tree.
COI gene analysis
The same set of analyses (ML, MP, NJ, and BI) was conducted using all available COI data. Final COI alignment comprised 661 bp (of which 227 bp, or 34.3%, were parsimony informative).
ML analysis was conducted generally as for the 16S sequences, except using a GTR+G+I model (General Time Reversible substitution model; discrete gamma distribution, G parameter = 1.051; allowing for invariant sites, 55.74% sites) with lowest BIC score = 9949.4. Gaps/missing data treatment was set to ‘partial deletion’, site coverage cut-off = 90% (resulting in 612 sites in the final dataset), and all codon positions and noncoding sites were considered in ML analysis (highest log-likelihood for initial replicate tree = −4524.3).
MP analysis of COI was as per the 16S MP tree, but included all codon positions and noncoding sites, again with site coverage cut-off = 90%, with 612 sites in final dataset (consistency index = 0.403, retention index = 0.756, composite index = 0.314 for all sites or 0.304 for parsimony-informative sites, most parsimonious tree length = 909).
NJ analysis was also as for the 16S NJ tree, but considering all codon positions and noncoding sites, and with the optimal tree with sum branch length = 1.216 (612 sites in final dataset).
BI analysis used the same GTR+G+I model as the ML analysis, and otherwise was conducted as per analysis of 16S.
P-distance calculations
For both 16S and COI sequencing data, the extent of sequence similarity and difference between and within putative family level clades (supported by molecular phylogenies) was calculated in order to partly assess robustness of different clades. Distance estimation used P-distance as the method/model (including transitions + transversions as substitutions), rates among sites was set as uniform rates, and gaps/missing data treatment was set to pairwise deletion. Average, minimum, and maximum distances were calculated within each group, and average distance was calculated between each group.
TimeTree analysis
To estimate cirrate clade divergence times, a timetree was constructed in MEGA X using the inbuilt RelTime-ML TimeTree estimation tool (Mello 2018). Because COI sequences had greater bp coverage than 16S sequences, greater percentage of parsimony informative sites (34% vs. 26%), and considerably greater tree log-likelihood values, COI data were used in timetree estimation.
The previously constructed ML phylogenetic tree with site-coverage cut-off 90% was used (supplying the COI alignment file and a Newick format export of the tree as inputs for the TimeTree tool). For TimeTree parameters the GTR model was used with discrete gamma distribution (G parameter = 1.299), and evolutionarily invariable sites (58.99% of sites). For timetree calibration, previous molecular clock analyses indicated Jurassic or Early Cretaceous (~210–110 Ma) divergence of the Incirrata from the Cirrata (Tanner et al. 2017). However, Fuchs & Schweigert (2018) hypothesised that Cirrata and Incirrata arose from the Patelloctopodidae Fuchs et Schweigert, 2018, represented by the genera Pearceiteuthis Hewitt et Jagt, 1999 (Callovian, 166–163 Ma) and Patelloctopus Fuchs et Schweigert, 2018 (Kimmerdigian, 157–152 Ma). Secondly, beaks of fossil Cirrata (Paleocirroteuthis Tanabe, Trask, Ross et Hikida, 2008) more closely resembling those of cirroteuthids than opisthoteuthids are known from the Cretaceous Santonian (86–84 Ma) and Lower Campanian (84–81 Ma) (Tanabe et al. 2008). Therefore, the node for the earliest division of Cirrata was given an upper bound of 164.5 Ma (mid-Callovian) when putative stem-cirrates were apparent, and a lower bound of 85 Ma (mid-Santonian) by which time cirrates resembling cirroteuthids had likely diverged from other cirrates. The calibrated node was set to ‘uniform distribution’, with the estimated log likelihood value of the resulting timetree being −4543.8.
RESULTS
PHYLOGENETIC ANALYSES
Phylogenetic trees (16S and COI) were generated covering all currently accepted cirrate genera and included sequences for type species of all accepted genera other than Grimpoteuthis.
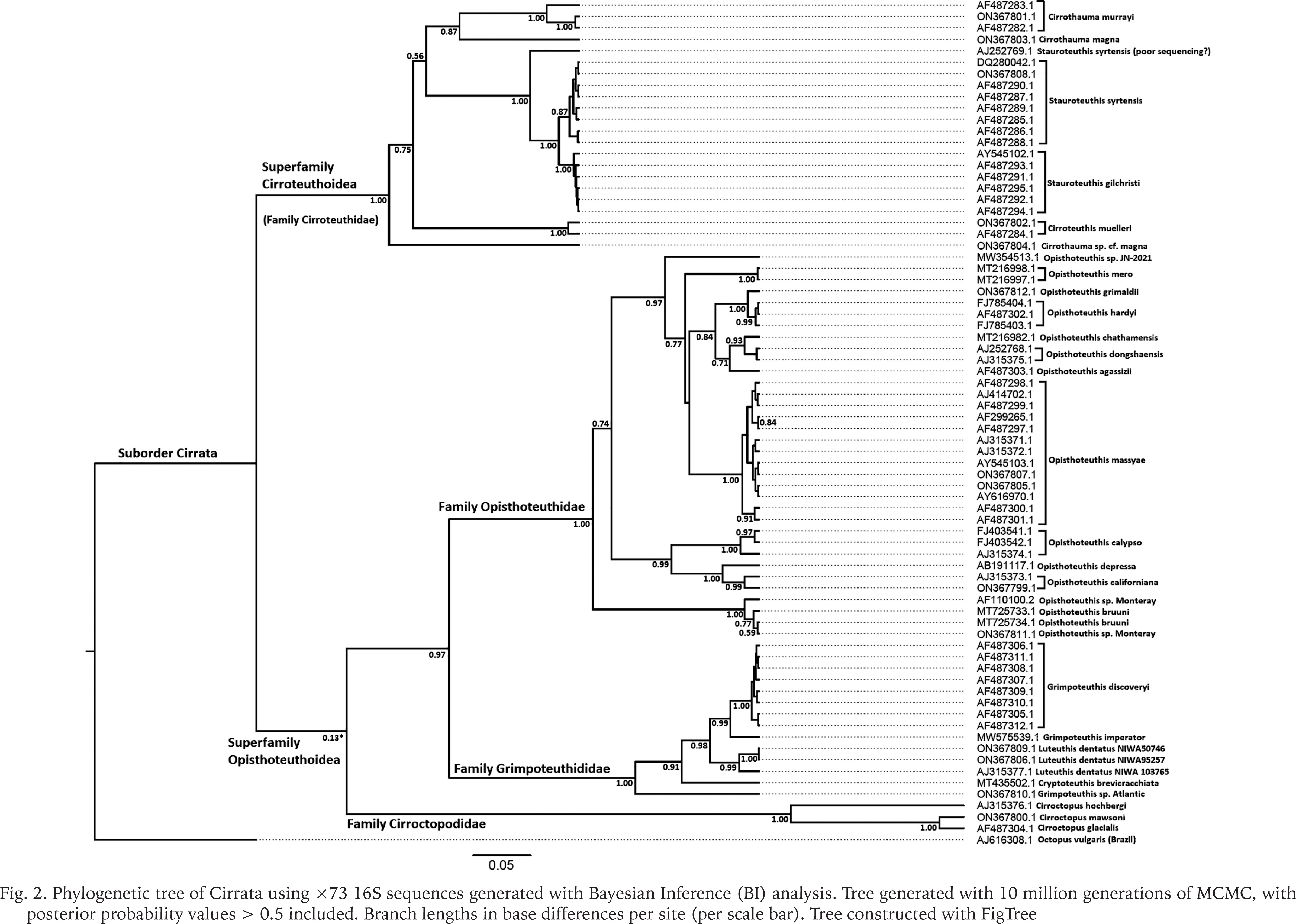
Phylogenetic trees using 16S covered a broader range of 26 putative species but had fewer parsimony informative sites and shorter sequences compared with COI (26% of 538 bp sites parsimony informative vs. 34% of 661 sites). The 16S phylogenetic analyses supported four monophyletic families, a basal-most Cirroteuthidae (including Stauroteuthis sensu Collins & Villanueva 2006) (bootstrap support 98% ML, 93% MP, and BI posterior probability (post-prob.) of 1.00), Cirroctopodidae (98–99% ML & MP, BI post-prob. 1.00), Grimpoteuthididae (72% ML, 93% MP, BI post-prob. 1.00), and Opisthoteuthidae (63% ML, 54% MP, Bi post-prob. 1.00). The phylogenetic trees for ML and MP are shown in Figure 1, BI tree in Figure 2. Within Cirroteuthidae, Stauroteuthis and Cirroteuthis formed clades (97–99% support in ML and MP, 1.00 BI post-prob.) within a consistently polyphyletic Cirrothauma. Stauroteuthis syrtensis Verrill, 1879 did not consistently separate from S. gilchristi (Robson, 1924) (though this may have been due to one S. syrtensis sequence being of poorer quality (AJ252769, and this sequence was excluded from later P-distance calculations). Other than this suspect S. syrtensis sequence, both S. syrtensis and S. gilchristi formed well supported clades, especially with BI (post-prob. values 0.87 and 1.00 respectively). Within Grimpoteuthididae, Grimpoteuthis was polyphyletic, with Grimpoteuthis sp. being sister to a clade containing Cryptoteuthis Collins, 2004, Luteuthis O’Shea, 1999, G. imperator Ziegler et Sagorny, 2021, and G. discovery Collins, 2003. The bootstrap support for Opisthoteuthidae in 16S phylogenies was lower with ML and MP analyses compared to other families, but still had a BI posterior probability of 1.00 (the same as other families). Within Opisthoteuthidae, species generally resolved as clades (many well supported). Interestingly, O. grimaldii (Joubin, 1903) and O. hardyi Villanueva, Collins, Sánchez et Voss, 2002(a) were very closely related (clade with 88–97% support in ML and MP, 1.00 BI post-prob.), but still resolved as likely separate species (O. hardyi forming a clade with 63–76% ML and MP support, 0.99 BI post-prob.) sister to O. grimaldii. Opisthoteuthis bruuni (Voss, 1982) (southeastern Pacific) and O. sp. ‘Monterey’ (Monterey Bay, northeastern Pacific) were polyphyletic within a well-supported clade, suggesting that they were conspecific, though, as with O. grimaldii and O. hardyi, some samples did form a well-supported sub-clade, indicating a need for further systematic assessment of eastern Pacific Opisthoteuthis. Higher level clades within Opisthoteuthidae were more strongly supported by the BI analysis of the 16S dataset, and two notable clades (BI post-prob. values 0.97–0.99) were one containing 3 species (O. calypso, O. californiana Berry, 1949, and O. depressa Ijima et Ikeda, 1895), and another containing all other species excluding O. bruuni / O. sp. ‘Monterey’.
Fig. 1
Phylogenetic tree of Cirrata using ×73 16S sequences as a combined result for Maximum Likelihood (ML) and Maximum Parsimony (MP) trees, both analyses with a site-coverage cutoff of 90%, tested with 1,000 bootstrap replicates. Topology is that of the MP tree, and bootstrap support values >50% are included next to clades in format ‘MP value/ML value’. Branch lengths measured in number of substitutions per site (per scale bar). Tree constructed with MEGA X
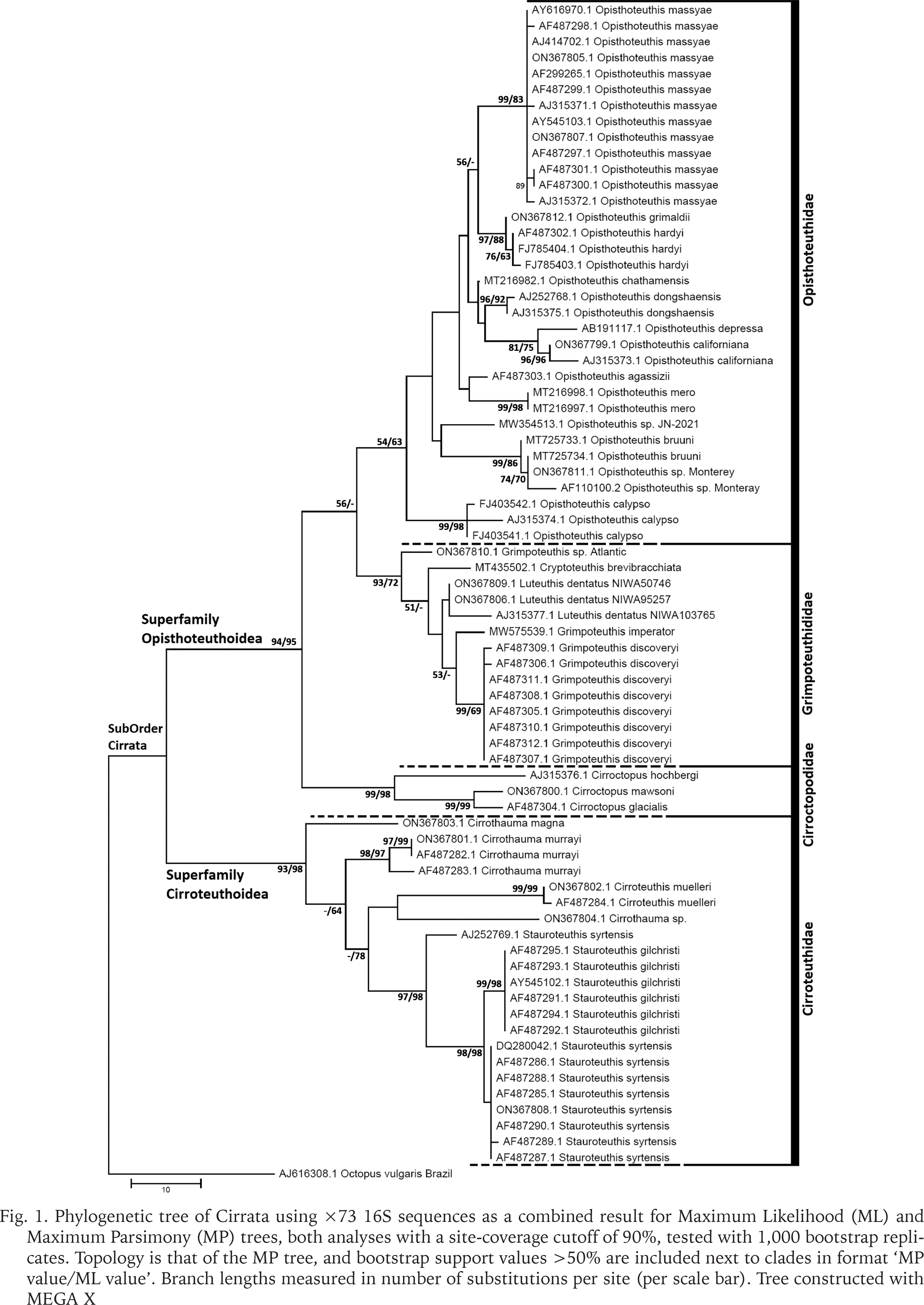
Fig. 2
Phylogenetic tree of Cirrata using ×73 16S sequences generated with Bayesian Inference (BI) analysis. Tree generated with 10 million generations of MCMC, with posterior probability values > 0.5 included. Branch lengths in base differences per site (per scale bar). Tree constructed with FigTree
Clade structure linking the Opisthoteuthidae, Grimpoteuthididae, and Cirroctopodidae varied, and was uncertain in MP and ML analyses (support values < 60%), while in BI analysis the Opisthoteuthidae and Grimpoteuthidae were more closely related (clade with 0.97 post-prob.). These three families formed a well-supported clade with 94–95% support in ML and MP, while in BI this clade had much lower support (0.13 post-prob).
The NJ tree for 16S data (not shown) produced a topology comparable to that of ML and MP trees, but with increased bootstrap support for many clades. The four families were resolved well: Cirroteuthidae (97%), Cirroctopodidae (99%), Grimpoteuthididae (98%), and Opisthoteuthidae (71%). The latter three families again resolved as a well-supported clade (97% support), and as with MP and ML analyses the family Opisthoteuthidae had lower support (though still > 70%).
Phylogenetic trees for COI had fewer sequences and covered only 18 taxa but included more parsimony-informative sites (ML and MP see Fig. 3, BI see Fig. 4). These trees produced similar topologies for families and genera, but with several interesting points of similarity and difference to the 16S trees.
Figs 3–4
Phylogenetic trees of Cirrata using ×41 COI sequences: 3 – combined results for ML and MP trees, site-coverage cutoff of 90%, tested with 1,000 bootstrap replicates, topology is that of the ML, and bootstrap support values >50% are included next to clades in format ‘MP value/ML value’. Branch lengths in substitutions per site (per scale bar). Tree constructed with MEGA X. 4 – BI tree generated with 10 million MCMC generations, and with posterior probability values > 0.5 included (values below this retained only for comparison and marked with ‘*’). Trees constructed with FigTree
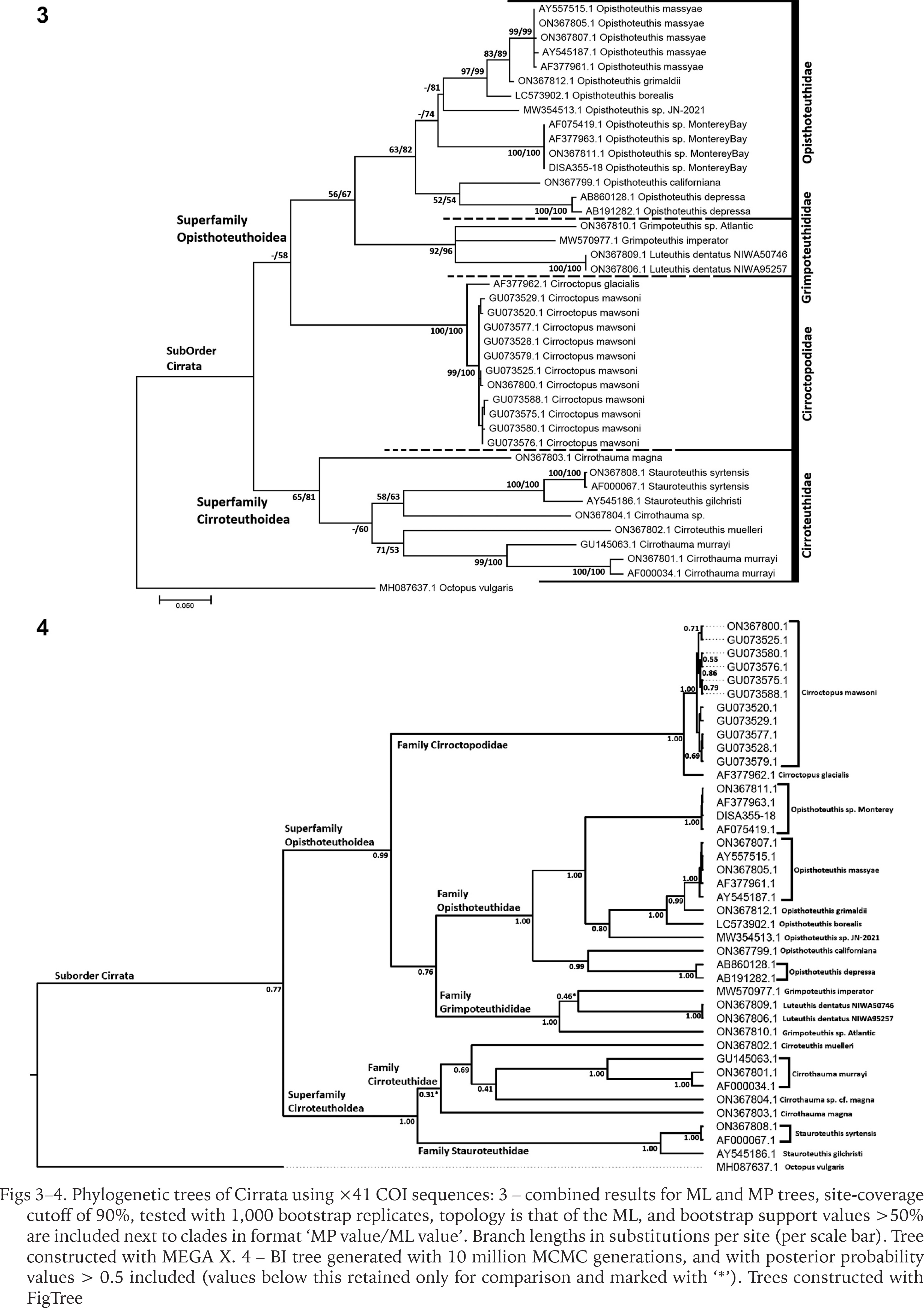
In the ML and MP trees, and NJ tree (not shown), Cirroteuthidae (bootstrap support 65–81%) again resolved as a clade basal to a more weakly-supported clade (55–58%) containing Cirroctopodidae (100% support), Grimpoteuthididae (92–96%), and Opisthoteuthidae (63–82%), the latter two also forming a clade with 56–67% support. Within Cirroteuthidae, Stauroteuthis again formed a clade (100% support), while it and Cirroteuthis were nested within a polyphyletic Cirrothauma. Notably, BI analysis resolved Stauroteuthis as a clade sister to one containing both Cirrothauma and Cirroteuthis, thus supporting two families Stauroteuthidae and Cirroteuthidae. However, while the former had high support (post-prob. 1.00) the latter had very low support (post-prob. 0.31), meaning the separation of these two families was only tentative. BI analysis again resolved Cirrata as two clades with high support (post-prob 0.99–1.00), one containing Cirroteuthidae (including Stauroteuthidae) and the other clade containing the other three families, Grimpoteuthididae, Opisthoteuthidae, and Cirroctopodidae, with these three families all having high BI post-prob. values of 1.00.
Unlike in 16S analyses, Luteuthis formed a clade (100% support, post-prob 1.00) but this was again within a polyphyletic Grimpoteuthis. Within Opisthoteuthis, and unlike in 16S analyses, COI MP, ML, NJ (not shown), and BI analyses consistently resolved clades of species within the genus. Opisthoteuthis depressa and O. californiana formed a weakly-supported clade (52–54% support, post-prob. 0.99) sister to a clade containing five other species (74% support, post-prob. 1.00), within which O. borealis Collins, 2005, O. grimaldii, and O. massyae formed a clade (97–99% support, post-prob. 1.00).
For both the 16S and COI datasets, it was apparent that additional (potentially undescribed) species were present. Notably in the polyphyletic Cirrothauma, three species C. magna (Hoyle, 1885), C. sp., and C. murrayi Chun, 1911 were all well separated from each other, and variable in placement between the different analyses. Even C. murrayi, while always forming a well supported clade (97–99% support or post-prob. 0.87–1.00, for 16S and COI analyses) was possibly comprised of two species, with one sequence appearing relatively distinct from the other two (see Figs 1–4). Overall, COI and 16S phylogenies suggest underappreciated species diversity in the Cirroteuthidae.
Within (intra)- and between (inter)-group genetic distances (P-distances; pairwise deletion) for the four putative families, for both 16S and COI data, were calculated and are presented in Table 4 (while Stauroteuthidae may have been apparent in the single COI BI analysis, this was the only analysis, and even in this the separation from Cirroteuthidae was weak, thus while Stauroteuthidae may be a valid family, it is not recognised herein). Inter-family genetic distances were consistently greater in COI compared to 16S, the greatest average inter-family genetic distance was for the Cirroteuthidae vs. Cirroctopodidae (15.5% 16S; 18.4% COI). The lowest average distance was between Opisthoteuthidae and Grimpoteuthididae (7.1% 16S; 14.2% COI), though this was comparable to the distance between Opisthoteuthidae and Cirroctopodidae (10.9% 16S; 15.1% COI) and Grimpoteuthididae and Cirroctopodidae (11.2% 16S; 15.9% COI). Average P-distances between Cirroteuthidae and Grimpoteuthididae (12.6% 16S; 18.0% COI) and Opisthoteuthidae (13.1% 16S; 18.3% COI) were between these.
Table 4
P-distances within and between putative family level clades for cirrates from 16S sequences (×72; 538 bp in final alignment) and COI sequences (×40; 661 bp in final alignment), removing all ambiguous positions (pairwise deletion). P-distances are converted to percentages
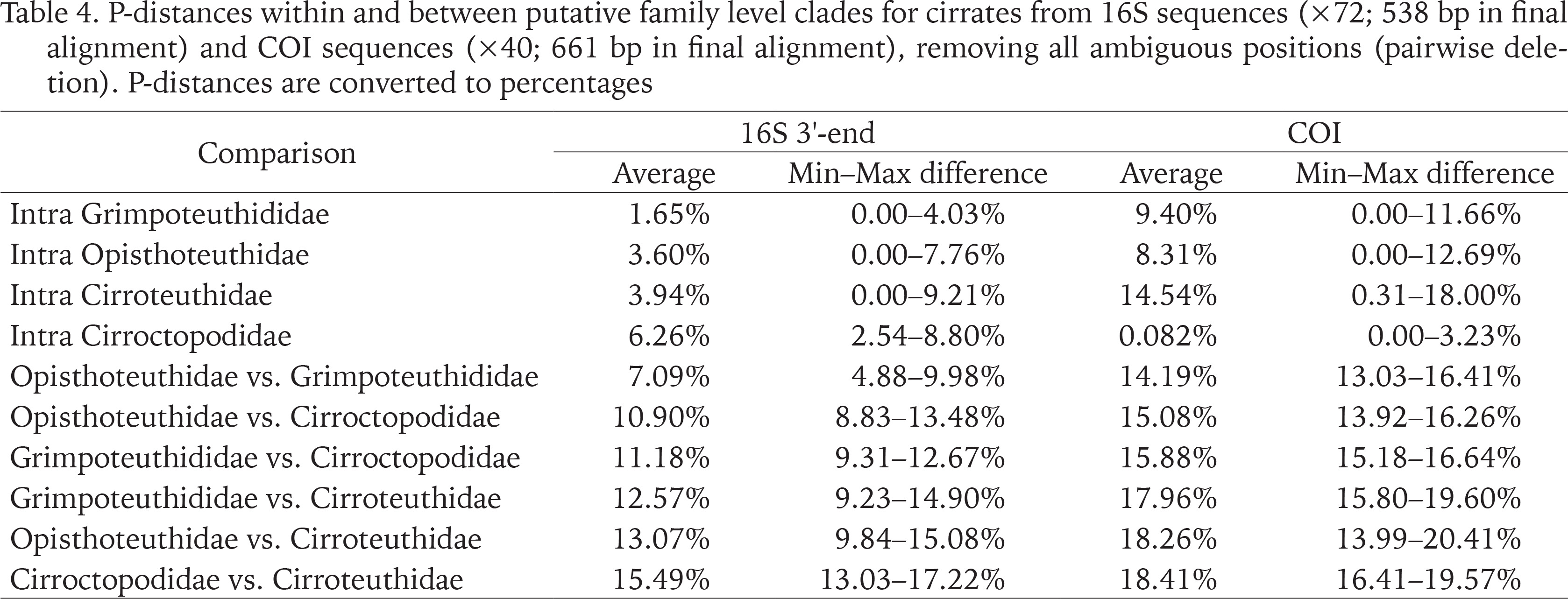
Intra-family P-distances were again higher for COI datasets compared to 16S (with the exception of Cirroctopodidae) (Table 4), and were consistently lower than inter-family distances, except for the intra-Cirroteuthidae distance for COI (14.5% average) which was comparable to the lowest inter-family P-distance for COI (giving some support for Stauroteuthidae as a distinct family – as indicated by the COI BI analysis). For 16S, genetic intra-family P-distances were lowest in Grimpoteuthididae (1.7% average), followed by Opisthoteuthidae (3.6% average), Cirroteuthidae (3.9% average), and Cirroctopodidae (6.3% average); whereas COI intra-family P-distances were lowest in Cirroctopodidae (0.8% average), followed by Opisthoteuthidae (8.3% average), Grimpoteuthididae (9.4% average), and Cirroteuthidae (14.5% average). Overall, inter- vs. intra-family P-distances were consistent with four families.
Systematic proposals
For both 16S and COI phylogenetic trees (across ML, MP, NJ, and BI trees), robust clades formed at the superfamily level, with Cirroteuthidae (including Stauroteuthidae) forming a sister clade to that containing other cirrate families. These two higher-level clades are consistently reported in other molecular studies that include Cirrata (Piertney et al. 2003, Sanchez et al. 2018, Pardo-Gandarillas et al. 2021, Ziegler & Sagorny 2021, Taite et al. 2023). Importantly, the morphology and lifestyle of these taxa also differ. Consequently, the following superfamily level clades are proposed, with the formal recognition of these superfamilies hopefully assisting understanding of cirrate evolutionary relationships and providing a framework for further study:
Superfamily Cirroteuthoidea nov.
Diagnosis. Cirrates with secondary (complex) webbing, elongate cirri (length > 4–10 × maximum sucker diameter), and sepioid gills.
Families. Cirroteuthidae (including Stauroteuthidae).
Remarks. Essentially pelagic or benthopelagic; spending the majority of time in the water column and only briefly contacting the seafloor for feeding (Collins & Villanueva2006, Golikov et al. 2023). Central arm sucker acetabular chamber reduced in Stauroteuthis, Cirroteuthis, and Cirrothauma, with some suckers elevated on fleshy stalks, and for Stauroteuthis at least, bioluminescent (Aldred et al. 1983, Johnsen et al. 1999, Verhoeff 2022). Gill lamellae counts of Stauroteuthis are lower than Cirroteuthis and Cirrothauma, and with increased lamellae branching, but they remain relatively elongate and sepioid (Young & Vecchione 2002).
Superfamily Opisthoteuthoidea nov.
Diagnosis. Cirrates without secondary webbing, with moderate to short cirri (length < 4× sucker diameter), and relatively compact gills (semi-sepioid or half orange form).
Families. Opisthoteuthidae, Grimpoteuthididae, and Cirroctopodidae.
Remarks. Primarily benthic, spending time resting on the seafloor, with occasional bursts of fin and medusoid swimming (Collins & Villanueva2006).
Divergence time estimations
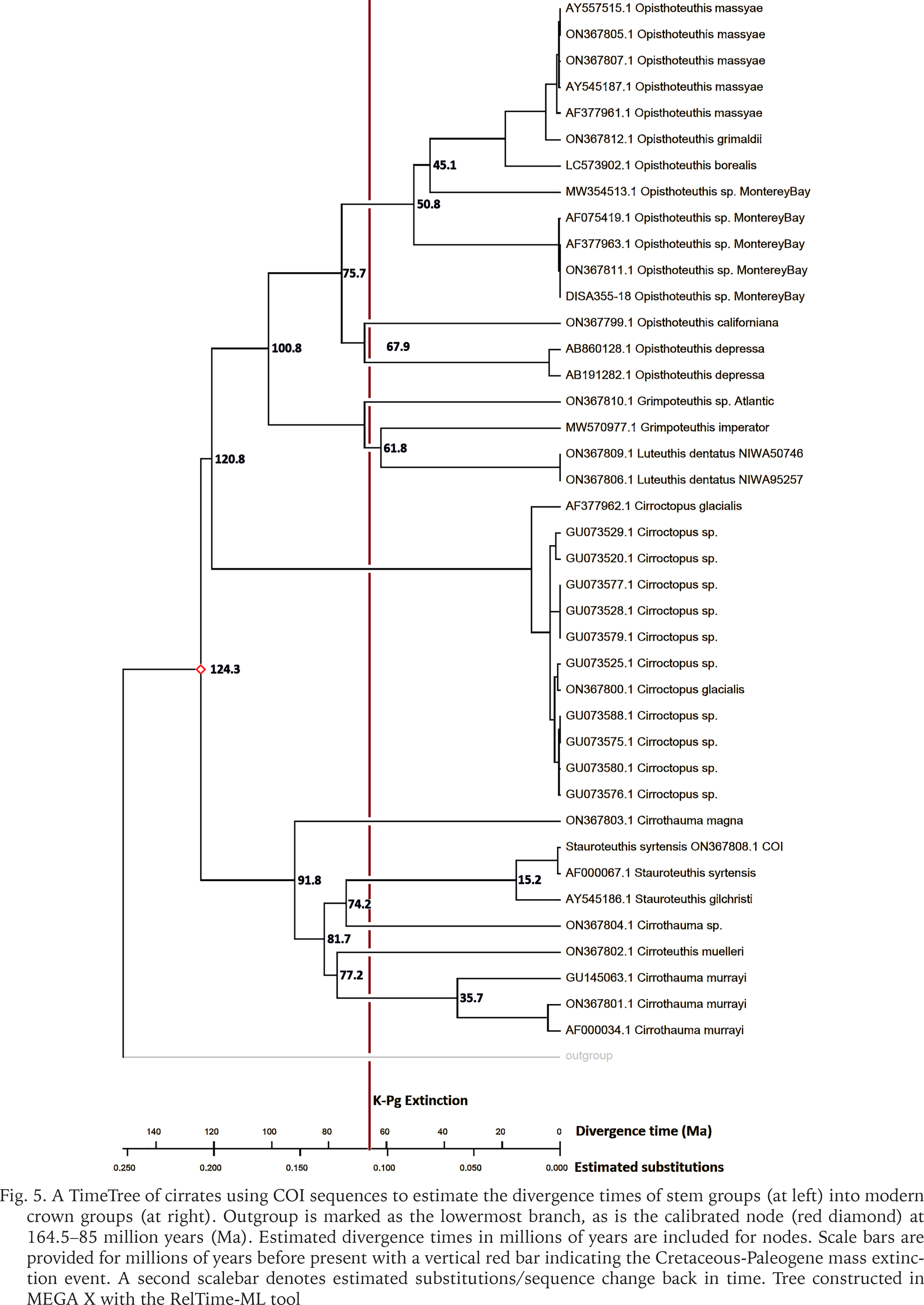
An investigation of cirrate divergence times was conducted by constructing a phylogenetic timetree from the COI sequencing data, calibrating the most-ancestral node between superfamilies Cirroteuthoidea and Opisthoteuthoidea at 164.5–85 Ma. The resulting timetree is depicted in Figure 5. This estimation (though limited by the scarcity of cirrate or stem-cirrate fossils) resulted in an estimated divergence of the Cirroteuthoidea and Opisthoteuthoidea at 124 Ma (Early Cretaceous). Within Opisthoteuthoidea, stem-cirroctopodids were estimated to have diverged at 121 Ma, and stem-grimpoteuthidids diverged from stem-opisthoteuthids at 101 Ma. The stem-Opisthoteuthis began diversifying in the Late Cretaceous (earliest node at 76 Ma), but most diversification of Opisthoteuthis is estimated to have occurred through the Paleogene (nodes within Opisthoteuthis having estimated times between 68–45 Ma). Within Cirroteuthidae, stem-Stauroteuthis, Cirroteuthis, and Cirrothauma diverged off from each other between 92–74 Ma (Late Cretaceous).
Fig. 5
A TimeTree of cirrates using COI sequences to estimate the divergence times of stem groups (at left) into modern crown groups (at right). Outgroup is marked as the lowermost branch, as is the calibrated node (red diamond) at 164.5–85 million years (Ma). Estimated divergence times in millions of years are included for nodes. Scale bars are provided for millions of years before present with a vertical red bar indicating the Cretaceous-Paleogene mass extinction event. A second scalebar denotes estimated substitutions/sequence change back in time. Tree constructed in MEGA X with the RelTime-ML tool
DISCUSSION
Recent decades have seen conflicting accounts of relationships between cirrate taxa being proposed (O’Shea 1999, Piertney et al. 2003, Collins & Villanueva 2006). The recent work by Taite et al. (2023) indicated that using many nuclear and mitochondrial genes could be good for interrogating higher-level relationships within octopods, including cirrates, however such sequencing is not yet available for the vast majority of cirrates. Molecular phylogenies using 16S (e.g., Piertney et al. 2003, Pardo-Gandarillas et al. 2021) have revealed comparable clade structure to that resolved by Taite et al. (2023) using whole mitochondrial/nuclear datasets for a much smaller number of species, indicating that smaller numbers of mitochondrial genes are still suitable for analysing higher family-level clades within Cirrata. Furthermore, 16S and COI sequences have the advantage of far greater coverage of species, allowing identification of paraphyletic genera, sub-genus structure, and putative new species (as is done herein). In future, it may be hoped that a much greater number of cirrate taxa will have large mitochondrial and nuclear gene sets sequenced, but this is unlikely to reach comparable species coverage to 16S and COI sequencing anytime soon. Up till now only a single mitochondrial gene (16S) has been widely used, and by combining a phylogenetic analysis of cirrate 16S sequences (including several species never used in such analyses before) with a large set of COI sequences which have never been thoroughly investigated for cirrate octopods, it was anticipated that the molecular phylogeny of Cirrata may be resolved to a far greater degree than possible before.
The most contentious of taxonomic points within cirrate taxonomy has involved the systematic validity of Grimpoteuthididae (whether it was distinct from Opisthoteuthidae), and Stauroteuthidae (whether it was distinct from Cirroteuthidae). Hochberg et al. (2014) and Vecchione et al. (2016) objected to the recognition of Grimpoteuthididae (synonymising it with Opisthoteuthidae) and synonymising of Stauroteuthidae, while later researchers accepted Grimpoteuthididae (and not Stauroteuthidae) (Ziegler et al. 2021) or accepted neither (Pardo-Gandarillas et al. 2021). Most molecular studies on Cirrata have relied exclusively on the 16S 3'-end sequence (Piertney et al. 2003, Pardo-Gandarillas et al. 2021), with the COI gene being rarely used despite its widespread use in metazoan barcoding (Barcode of Life project or BOLD; Ratnasingham & Herbert 2007) and identification of cephalopod species (e.g., Gebhardt & Knebelsberger 2015). The first relatively comprehensive molecular phylogeny of cirrates based on the COI gene found support for Cirroteuthidae, Stauroteuthidae, Opisthoteuthidae, and Cirroctopodidae (Golikov et al. 2020); an earlier COI tree of Carlini et al. (2001) was based on only five taxa. Because COI sequences for Cirroteuthis or grimpoteuthidids (other than a species of Opisthoteuthis erroneously attributed to Grimpoteuthis, discussed later) were unavailable to Golikov et al. (2020), their COI phylogeny could not resolve the monophyly of Cirroteuthidae or validity of Grimpoteuthididae. However, the accumulation of 16S and COI sequence data now permits a much more comprehensive assessment of clade structure using both these mitochondrial genes.
Grimpoteuthididae
Well-supported clades consistent with four families (Cirroteuthidae, Cirroctopodidae, Grimpoteuthididae, and Opisthoteuthidae) were apparent in both 16S and COI analyses (bootstrap values > 90% or post-prob. values approaching 1.00). Opisthoteuthidae was occasionally more weakly supported in ML/MP, but strongly supported in BI. Such support values were consistent with family level support reported by Piertney et al. (2003), but support herein was much stronger for Grimpoteuthididae. Similarly, support across ML and BI analyses for the families was consistent with values reported by Pardo-Gandarillas et al. (2021) (though these authors did not recognise Grimpoteuthididae, regardless of its high support and did not include Cryptoteuthis). While Grimpoteuthididae and Opisthoteuthidae had an average 16S inter-clade P-distance of 7.1%, this difference exceeded that of 16S intra-family genetic distances (including Opisthoteuthidae (3.6%) and Grimpoteuthididae (1.7%)) and was comparable to 16S P-distances between Cirroctopodidae relative to both Opisthoteuthidae and Grimpoteuthididae (10.9% and 11.2%, respectively). COI P-distances between families, which included more parsimony informative sites, provided even greater support for separation of these families, with values between Opisthoteuthidae, Grimpoteuthididae, and Cirroctopodidae ranging from 14.2–15.9%, again far exceeding COI intra-family distances for these three families (0.8–9.4%). While Grimpoteuthididae and Opisthoteuthidae often formed a clade sister to Cirroctopodidae, in some 16S analyses (when using a lower site-coverage cut-off) this clade structure is reversed, and Cirroctopodidae and Opisthoteuthidae formed a clade to which Grimpoteuthididae was sister (see also Ziegler & Sagorny 2021). Similarly, using 18S and 28S nuclear genes, Taite et al. (2023) reported relationships between these three families to be unclear (i.e., a polytomy). Based on morphology, Cirroctopodidae and Opisthoteuthidae appear to be closely related, in that they share branched optic nerve bundles through the white body, have internal shells with tapering wing-ends, and lack a radula and posterior salivary glands. Whereas Grimpoteuthididae differ greatly in having a single optic nerve bundle through each white body (a feature shared with Cirroteuthidae and Stauroteuthidae), internal shells with expanded wing ends, and frequently possess a radula (Collins & Villanueva (2006); and personal observations).
Overall, based on clade bootstrap support, comparable inter-family P-distances (especially with COI) between grimpoteuthidids, opisthoteuthids, and cirroctopodids relative to low intra-family P-distances, and distinct morphological differences, Grimpoteuthididae is at least as valid a family as is the Cirroctopodidae; its synonymy with Opisthoteuthidae is not supported on either morphological or molecular grounds.
Cirroteuthidae – monophyly and new taxa
Herein 16S and COI sequences were analysed for all five recognised cirroteuthid species for the first time (genera Cirroteuthis, Cirrothauma, and Stauroteuthis). Consistent with earlier molecular analyses, the Cirroteuthidae is paraphyletic, in that it contains the mono-generic Stauroteuthidae (i.e., genus Stauroteuthis) (Piertney et al. 2003, Pardo-Gandarillas et al. 2021, Ziegler & Sagorny 2021). Accordingly, there is little 16S or COI molecular support to recognise the Stauroteuthidae as discrete from the Cirroteuthidae. Cirrothauma is consistently resolved as paraphyletic, although the exact positioning of taxa relative to each other differs in 16S and COI trees. This indicates that further morphological study of this genus is warranted. Furthermore, Cirrothauma sp. sequence (ON367804) from the northeastern Pacific (Gorda Ridge) is well separated from C. magna (Hoyle, 1885) and C. murrayi Chun, 1911 in both 16S and COI trees (resolving as closer to one or the other depending on the analysis), indicating the existence of at least one more Cirrothauma species in the northern Pacific. Unfortunately, the ‘known’ C. magna sequence (ON367803) is from the North Atlantic, not the type locality in the southern Indian Ocean (though the type locality of the ‘nomen dubium’ Cirrothauma hoylei (Robson, 1932) is closer, in the southern Pacific)1; to determine the sequence of ‘true’ C. magna, and assess if C. hoylei may be an appropriate name for this third Cirrothauma species, will require an integrated molecular and morphological analysis of material from or at least proximal to the type locality. Cirrothauma murrayi COI sequences from the North Atlantic (ON367801 & GU145063) and North Pacific (AF000034) resolve as a clade, but the Pacific specimen is sister to the others, and P-distances between these three sequences (8.0%) are greater that within other widely distributed species (e.g., COI sequences for O. massyae from north Atlantic and off South Africa had an intra-species P-distance = 0.3%), indicating that C. murrayi may comprise more than one species rather than a the single cosmopolitan species reported by Aldred et al. (1983). Cirroteuthids as a family are generally poorly studied, and in addition to the existence of a third (or fourth) species of Cirrothauma supported herein, new species of Cirroteuthis are likely (Verhoeff 2022), and a new species of Stauroteuthis from the southwestern Pacific was recently described (Verhoeff 2023).
The molecular work of Taite et al. (2023) indicated that relationships between Cirroteuthis, Cirrothauma, and Stauroteuthis may be better resolved with nuclear 18S and 28S rRNA genes. In their ML phylogeny, S. syrtensis was sister to a well-supported clade containing Cirroteuthis and Cirrothauma (with Cirroteuthis in turn a sister taxon to a clade containing the three Cirrothauma species). While most analyses of 16S and COI herein failed to recover Stauroteuthidae as a family separate to Cirroteuthidae, this was found in a single analysis (BI analysis of COI, see Fig. 4), though the post-prob. for Cirroteuthidae as a sister clade to Stauroteuthidae was unusually low. Thus, the COI gene may provide better resolution of cirrate family structures than 16S, though only marginally, and nuclear rRNA genes support the separation of the Stauroteuthidae and Cirroteuthidae somewhat better (but these genes are non-sequenced for most cirrate taxa). Regardless, molecular support for considering the Stauroteuthidae as a valid family remains more tenuous than that for recognising the Grimpoteuthididae as a valid family, and recognition of Stauroteuthidae as a family must rely more heavily on morphological differences, notably the U-shaped internal shell which is distinct from the saddle- or butterfly-shaped shell of Cirroteuthis and Cirrothauma (Collins & Villanueva 2006).
New superfamily divisions of Cirrata
At a higher taxonomic level, multigene phylogenies of cephalopods have revealed monophyly of the Cirrata and Incirrata (Lindgren et al. 2012, Taite et al. 2023), as well as potential paraphyly, with Cirroteuthidae (including Stauroteuthidae) forming a basal sister clade to Incirrata and the remaining Cirrata (Carlini et al. 20012, Sanchez et al. 2018) (though paraphyly was doubted by the study authors). This reflects findings presented herein insomuch as the cirroteuthids (including Stauroteuthis) form a distinct clade sister to that containing other cirrate families in both 16S and COI phylogenies (bootstrap and post-prob. support for these two clades is consistently high for 16S and COI). These two clades are also reported in most molecular studies of Octopoda (Piertney et al. 2003, Lindgren et al. 2012, Sanchez et al. 2018, Pardo-Gandarillas et al. 2021, Taite et al. 2023). The consistent phylogenetic separation and strong morphological differences between these two higher-level clades supports the designation of them as superfamilies, the Cirroteuthoidea nov. (cirroteuthids with secondary webbing, very-elongate cirri, and sepioid gills) and Opisthoteuthoidea nov. (opisthoteuthids, grimpoteuthidids, and cirroctopodids, principally characterised by having a simple web, relatively short cirri, and half-orange/semi-sepioid gills).
Opisthoteuthidae & Grimpoteuthididae – new species and paraphyletic genera
Presently unidentified, potentially new species are apparent within Opisthoteuthis molecular phylogenies. The northwestern Pacific Opisthoteuthis sp. ‘JN-2021’ (MW354513) could belong to an already described species, but other North Pacific species (O. depressa, O. californiana, and O. dongshaensis are already represented, though the validity of species assignments to some of these sequences must be questioned). The northeastern Pacific (off Los Angeles and Monterey Bay) Opisthoteuthis sp. ‘Monterey’ – a taxon referred to as Grimpoteuthis sp. (Hunt 1999, Norman 2000: p. 182) and informally as O. ‘adorabilis’ (Baker, 2015) – forms a well-supported clade, but in the 16S datasets this clade includes sequences for O. bruuni, indicating that the two are possibly conspecific or a closely related species complex. Prior to Collins (2003), O. bruuni was placed in Grimpoteuthis (Voss 1982). Hochberg et al. (2014: p. 250) further remarked “… a similar [to O. bruuni], or closely related, undescribed species, occurs in the northeastern Pacific Ocean off California.” The attribution of these northeastern Pacific specimens to O. bruuni represents a considerable expansion of the recognised distribution of this species (from Chile to Monterey Bay). However such a range is not unusual in Opisthoteuthis, and this distribution is comparable in latitudinal extent to the Atlantic ranges of O. massyae and O. calypso (from the North Sea and Mediterranean to southern Africa) (Villanueva et al. 2002b).
Regarding other Opisthoteuthis, AJ252768 (tentatively assigned O. dongshaensis) was from a specimen supposedly collected off California (northeastern Pacific), but it forms a clade with a known O. dongshaensis sequence (AJ315375) from a specimen collected off Taiwan (Hudelot 2000, Piertney et al. 2003). Either O. dongshaensis is distributed more widely than hitherto recognised, or the locality data or identification of one or both samples is confused.3
COI barcoding sequences confer advantages over 16S 3'-end sequences for resolving cirrate octopod phylogenies, in that the area amplified contains more parsimony informative sites (34% vs. 26% in 16S sequences). COI may be more appropriate for investigating intra-genus structure within Opisthoteuthis (or perhaps sub-family or new genus divisions within Opisthoteuthidae) and COI bootstrap support values were higher for clades within Opisthoteuthis (compared to 16S). The 16S analysis herein, but only the BI analysis, also revealed some well supported clades as described in results. The species coverage unfortunately differed between COI and 16S analyses, but the relatedness of O. massyae and O. grimaldii in a well-supported clade was consistent in both. The only other study that published a molecular phylogeny of Opisthoteuthis COI sequences only included four species with no support values (Golikov et al. 2020), while subgenus structure within Opisthoteuthis in 16S analyses herein differed considerably from that revealed by Pardo-Gandarillas et al. 2021 (though their phylogeny was far less complete, lacking O. agassizii, O. grimaldii, O. dongshaensis, and O. sp. ‘Monterey’). Wakabayashi et al. (2012) similarly found that COI was better at resolving genus-level and inter-subfamily level differences in Ommastrephid squids. Collection of COI sequences from more Opisthoteuthis species may identify support for new genera or subgenera; O’Shea (1999: pp. 16–34) recognised three Opisthoteuthis ‘groups’, the systematic validity of which have not been further investigated.
While grimpoteuthidids form a robust clade, Grimpoteuthis is paraphyletic (especially in 16S-derived phylogenetic trees) with Cryptoteuthis and Luteuthis being nested within three different Grimpoteuthis species. The identity of the Grimpoteuthis sp. sequence (ON367810) is intriguing in that the specimen was collected from the northeastern Atlantic. Because this sequence is distinct from that of G. discovery, it may belong to any of G. boylei Collins, 2003, G. challengeri Collins, 2003, G. wuelkeri (Grimpe, 1920), or G. umbellata (Fischer, 1884), that have all been described from this region. As discussed in Verhoeff & O’Shea (2022), Grimpoteuthis may comprise several genera, but the poor condition of the type specimen of G. umbellata, the type species of the genus, precludes a detailed comparison. While both Enigmatiteuthis O’Shea, 1999 and Cryptoteuthis may be available names for species currently grouped in a paraphyletic Grimpoteuthis, re-allocating species to either genus (or defining new genera) is premature until G. umbellata is redescribed based on material collected from or proximal to the type locality. Grimpoteuthidids are also greatly underrepresented in sequence data, represented by only 4 of 20 species. The nesting of a monophyletic Luteuthis clade within a polyphyletic Grimpoteuthis is remarkable considering how distinct Luteuthis is morphologically from the species classed as Grimpoteuthis (and Cryptoteuthis). While Luteuthis shares some features in common with other grimpoteuthidids (single optic nerve bundle, internal shell with expanded wing ends, and retained radula) it is readily distinguished by possessing a bilobed digestive gland, more radular teeth per transverse row, palatine teeth on labial palps, extremely short cirri, and strongly crenulated suckers (O’Shea 1999, O’Shea & Lu 2002, Collins & Villanueva 2006). While morphologically distinct, the positioning of Luteuthis within otherwise (superficially) similar taxa of Grimpoteuthis (and Cryptoteuthis) indicates that the family Luteuthididae cannot be supported as distinct from Grimpoteuthididiae, but moreover emphasises how unrelated many Grimpoteuthis species likely are from one another and how the diversity in this family has been overlooked, perhaps due to evolutionary homeomorphy in the family.
Divergence times of Cirrata
The molecular clock analyses of Tanner et al. (2017) indicate that crown coleoids diverged into octopodiforms and decapodiforms in the Late Carboniferous or Permian, with the divergence of stem-Octopoda into the Incirrata and Cirrata occurring within the Jurassic between ~210 Ma and 150 Ma (using CIR clock model). Stem-Octobrachia (superorder) comprised three suborders (Prototeuthidina, Loligosepiina, Teudopseina) in the Mesozoic (Fuchs & Weis 2008, Fuchs & Larson 2011a, b), with a robust internal shell, as well as eight arms and cirri (at least in the latter two) being an ancestral condition reflected in the Cirrata. Fossil and recent vampyroteuthids (Vampyronassa Fischer et Riou, 2002 and Vampyroteuthis, respectively) (Rowe et al. 2022), which likely arose from within Loligosepiina (Fuchs & Weis 2008), also share these ancestral conditions (i.e., cirri and robust internal shell) with Cirrata. The suborder Teudopseina, abundant and diverse within the Jurassic and Cretaceous, likely contains the stem-Octopoda (pre-Cirrata–Incirrata common ancestor) based on gladius morphology. Teudopseids including Glyphiteuthis Reuss, 1854 (Trachyteuthididae) had eight arms equipped with cirri (Fuchs & Larson 2011a), with teudopseids of family Patelloctopodidae (superfamily Muensterelloidea) including Pearceiteuthis from the Jurassic (Callovian 166–163 Ma) and Patelloctopus (Kimmerdigian 157–152 Ma) possibly comprising stem-Octopoda, as they display clear reduction and shortening of the median field (restricting the gladius vestige to the mid–posterior mantle alike Cirrata) while retaining expanded lateral fields for fin muscle attachments (Fuchs & Schweigert 2018). By the Late Cretaceous (Santonian to lower Campanian, ~86–81 Ma) cirrate octopods were already present in the fossil record with Paleocirroteuthis haggarti Tanabe, Trask, Ross et Hikida, 2008 and P. pacifica Tanabe, Trask, Ross et Hikida, 2008 beaks resembling modern cirroteuthids (Tanabe et al. 2008). Likewise, other Late Cretaceous species including Palaeoctopus newboldi (Woodward, 1896), Keuppia levante Fuchs, Bracchi et Weis, 2009, and K. hyperbolaris Fuchs, Bracchi et Weis, 2009, while still bearing paired mantle fins (at least in Palaeoctopus), had a posterior separation of the gladius into two parts, indicating that stem-Incirrata were already diversified (Fuchs et al. 2009). Molecular timetree analysis presented herein indicates that the Cirrata diverged into its basal-most clades ~124 Ma (with stem-cirroteuthids diverging), within the Early Cretaceous, followed by the rapid divergence of stem-cirroctopodids, grimpoteuthidids, and opisthoteuthids (and most genera) within the remainder of the Early and Late Cretaceous.
Tanner et al. (2017) estimated that the incirrate octopods diversified in the mid–late Cretaceous, with stem Decabrachia diversifying into sepiids, and mypopsid and oegopsid squid from the Jurassic to Cretaceous. This diversification was likely part of the Mesozoic Marine Revolution (MMR) (sensu Vermeij 1977), a period of enhanced predator–prey coevolution and ecological shifts over the Jurassic and Cretaceous, possibly furthered by expansion of the Atlantic and Indian Ocean basins in the Early Cretaceous. The herein predicted diversification of Cirrata within the Cretaceous (~124–66 Ma) may also have been part of the MMR. While cirrates and vampyroteuthids are today essentially restricted to the deep sea, late-Mesozoic cirrates likely diversified in shallower shelf waters and were plausibly more active. Unalike modern zooplanktivorous and deep-sea-dwelling Vampyroteuthis, fossil vampyrotethids such as Vampyronassa rhodanica Fischer et Riou, 2002 were active predators of shallower regions (Rowe et al. 2022), while potential stem-Octopoda (Cirrata–Incirrata ancestor) Patelloctopus were collected from shallow marine deposits (< 100 m depth) (Fuchs & Schweigert 2018). At the extinction event at the end of the Cretaceous, cirrates and other cephalopods likely survived in the deep-sea (Arkhipkin et al. 2012), and while squid and Incirrata re-colonised shallower waters and diversified, the Cirrata and vampyroteuthids did not.
CONCLUSIONS AND FUTURE DIRECTIONS
The Cirrata comprises four well-supported families within two superfamilies, Cirroteuthoidea (for the Cirroteuthidae), and Opisthoteuthoidea (for the Cirroctopodidae, Grimpoteuthididae, and Opisthoteuthidae). While most cirrate genera form clades, Grimpoteuthis and Cirrothauma are polyphyletic. An integrated morphological and molecular approach is required to assess putative additional Cirrothauma and Opisthoteuthis species revealed herein, including assessing if historical names (including supposed junior synonyms) are applicable or if these are species new to science, and compare preserved specimens to more-recently collected, presently unidentified material. While COI sequences may be more informative for revealing cirrate phylogenies and taxonomy (because they contain more parsimony informative sites), more sequences for more species, especially the under-represented Grimpoteuthididae, are required. Future directions of study suggested by this research include: 1) systematic review of genera Grimpoteuthis, Cryptoteuthis, and Enigmatiteuthis with redescription of Grimpoteuthis type species G. umbellata using new material; 2) sequencing both COI and 16S for a greater number of grimpoteuthidid taxa (though sequencing larger sets including nuclear genes when practical); 3) redescription of North Pacific Opisthoteuthis species which may have been historically confused; and 4) increased collection of COI sequences from Opisthoteuthis taxa to assess sub-genus structure.